


1. Introduction
Selective Immunoglobulin M deficiency (SIgMD) is a predominantly antibody-affecting deficiency that has been included in the International Union of Immunological Societies (IUIS) classification of inborn errors of immunity (IEI) since 2017 [1,2,3]. According to the European Society for Immunodeficiencies (ESID) criteria, SIgMD is defined by absent or reduced serum immunoglobulin M (IgM) levels in the absence of immunoglobulin deficiencies of the other classes [4]. The significant comorbidities for SIgMD are recurrent infections (even including those with life-threatening severe infections) and an increased frequency of allergic and autoimmune diseases [5]. However, only a few studies have assessed the clinical and immunological features of SIgMD. The pathogenesis of SIgMD remains unclear, and no definitive genetic alterations have been established. SIgMD remains a diagnostic and therapeutic dilemma, especially in the pediatric population, because no conclusive data are available on the correct therapeutic management and the prognosis of SIgMD.
This review aims to summarize the evidence about this apparently rare primary immunodeficiency (PID), focusing on clinical and immunological features, particularly in pediatric patients, to allow for a better understanding of this condition.
2. Materials and Methods
A literature search was performed via the online database PubMed by combining the terms “Primary selective IgM deficiency”, “Primary selective immunoglobulin M immunodeficiency”, “Selective IgM deficiency AND children”, “Selective IgM deficiency AND pediatric population”, “Pediatric selective IgM immunodeficiency”, and “gamma-M deficiency AND children”.
The literature review on SIgMD was performed in May 2023, including all publication years. All studies that met the following criteria were included: (i) articles published in English in peer-reviewed journals and (ii) studies wherein the participants were children and adult patients diagnosed with SIgMD. Potentially eligible publications were manually screened and reviewed, and non-relevant publications were excluded.
3. Results
The database search found 850 articles. Based on their titles and abstracts, 166 articles met the inclusion criteria. After removing duplicates, 80 articles were analyzed for this review (Figure 1).
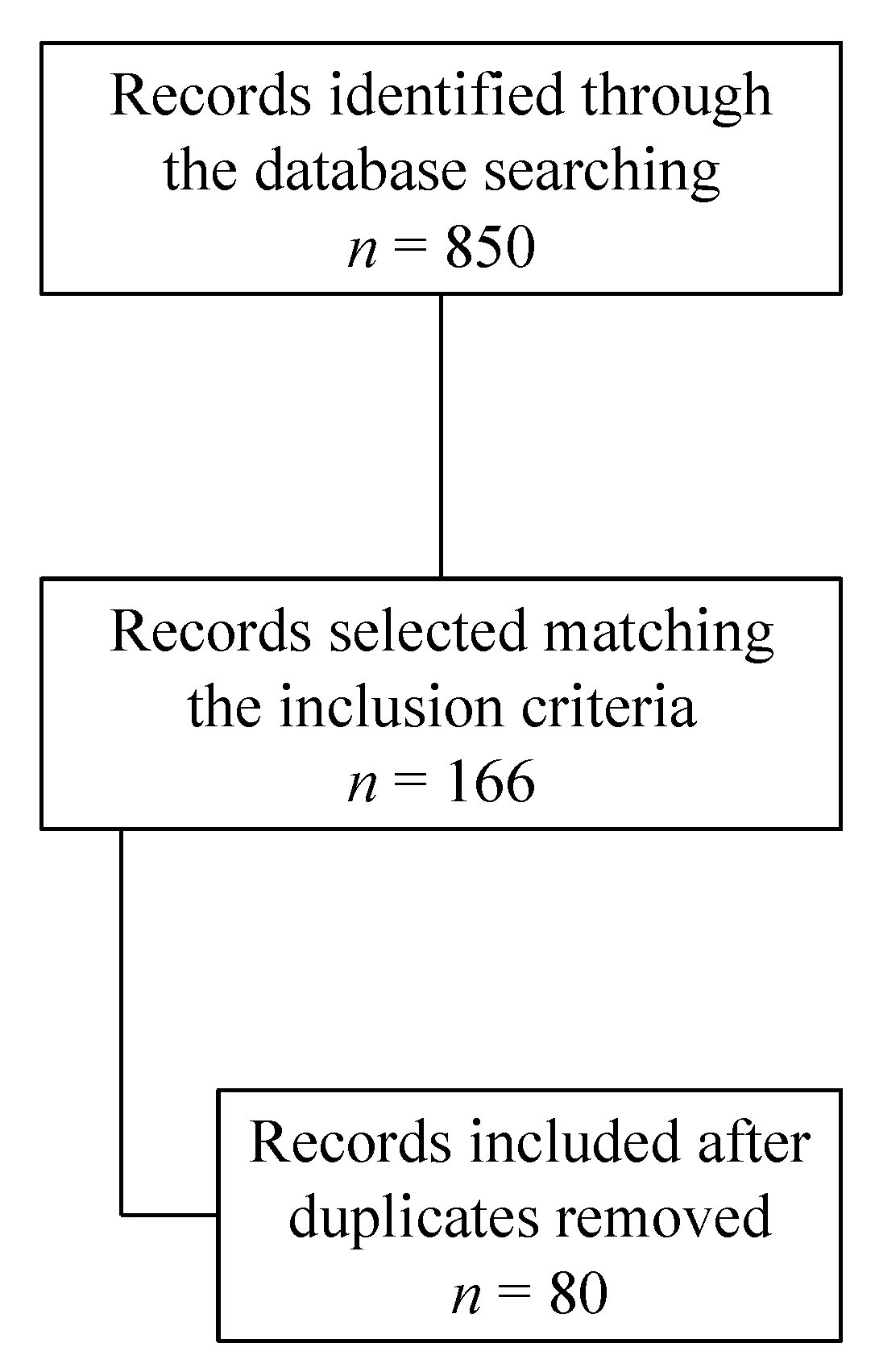
Figure 1. Search strategy.
3.1. Definition (Clinical and Laboratory)
According to the European Society for Immunodeficiencies (ESID) registry criteria, SIgMD is defined as repeatedly absent or reduced serum immunoglobulin M (IgM) levels (less than 2 SD or <10% of the values obtained from healthy controls of the same age or an absolute value <20 mg/dL in pediatric age) with normal levels of serum immunoglobulin A (IgA) and immunoglobulin G (IgG) and IgG subclasses, normal vaccine responses, and the absence of T cell defects (numbers and function) after the exclusion of secondary hypogammaglobulinemia (infections, genetic syndromes, chromosomal abnormalities, drugs, lymphomas, protein-losing enteropathy, nephrotic syndrome, thymoma) [6,7,8] and any other specific IEI. Despite this definition, some authors propose that SIgMD should be defined without the exclusion of the IgG subclass deficiency, alterations in T cell subset numbers and functions, and impaired responses to vaccines in order to better understand the different clinical and immunological phenotypes that lie behind the diagnostic term “IgM deficiency” [9]. In this context, in their 2022 Update of the Classification, the International Union of Immunological Societies (IUIS) Expert Committee defines the only main criteria as the absence/reduction in serum IgM without limiting the definition with additional features [2].
3.2. Epidemiology
SIgMD was described for the first time in 1967 by Hobbs et al. in children presenting with meningococcal meningitis [10], and it was considered to be a rare condition [11,12,13,14,15]. It is still mostly ignored as an IEI [16], and no large-scale studies have reported its epidemiology. An unselected community health screening survey reported a prevalence of 0.03% of patients with a complete absence of IgM [17]. More recently, in a screening of more than 3000 healthy adult blood bank donors in Iran, the prevalence of SIgMD was 0.37% [18]. A 0.07–2.1% prevalence in Immunology Clinics has been reported [19]. However, the prevalence of SIgMD in the pediatric population (<18 years of age) is unknown. In children, the reported median age at the onset of symptoms is 3 years, with a median age of diagnosis of 8 years [20] and an average age at the time of diagnosis of 6.0 ± 4.7 years [21]. Male predominance has been reported, along with a variable ratio of male to female patients (7:5 [21], 3:1 [20]).
3.3. Etiopathogenesis and Pathophysiology
3.4. Clinical Manifestations (Table 4)
Recurrent infections represent the presenting manifestation in more than 80% of patients with SIgMD [5,42,43]. Upper respiratory tract infections (including rhinitis, otitis media, and sinusitis) and pneumonia (also recurrent with the possibility of developing bronchiectasis [44]) represent the most common clinical manifestations in SIgMD patients [45,46]. However, invasive infections like septic arthritis [47] and severe life-threatening infections (bacterial meningitis and sepsis) have been reported [43,48,49,50]. Some of the most common microbial organisms include Streptococcus pneumoniae, Hemophilus influenzae, Neisseria meningitidis, Pseudomonas aeruginosa [48], Aspergillus fumigatus, and Giardia lamblia [51]. In children, infectious agents are also represented by Pneumocystis carinii, S. aureus, Salmonella sp, CMV, and Molluscum contagiosum [52].
Mycobacteria infections have also been described in association with SIgMD [46]. Hassanein et al. identified a case of miliar tuberculosis in a 31-year-old man [53]. Of note, as reported by Consonni et al., SIgMD may be associated with a severe clinical course of Mycobacterium infection compared to other healthy children [54]. Other infectious manifestations, such as skin infections [20,21,55,56,57], multiple recurrent hordeola [58], chronic gastritis, cholecystitis, and epididymitis [15], have been reported in these patients.
Several patients (up to almost 40% of patients with SIgMD) display allergic manifestations [20,21], and the frequency of asthma and allergic rhinitis in SIgMD ranges from 30 to 45% [5]. Likewise, autoimmunity and autoimmune diseases are more frequent in patients with SIgMD than in the general population [59].
Goldstein et al. reported autoimmune diseases in 14% of patients with SIgMD [19], although, in children, they are less frequent [46]. Autoimmune diseases, including systemic lupus erythematosus [60], Hashimoto thyroiditis [61], autoimmune thrombocytopenia [62], autoimmune glomerulonephritis [62], autoimmune hepatitis [63], juvenile idiopathic arthritis [64], and rheumatoid arthritis have been described as comorbidities in SIgMD. Also, chronic recurrent multifocal osteomyelitis [65], rheumatic heart disease, psoriasis, and scleroderma [15] have been reported. Lim et al. reported lupus-like nephritis and proteinuria in a 6-year-old patient with a mutation in the BTK gene [29]. Like other IEIs, hemophagocytic lymphohistiocytosis (HLH) is reported to be a potential complication of SIgMD [66].
Some cases of neoplastic disease have been reported, particularly in the adult population, while in children, neoplastic diseases are anecdotal [20,67,68,69,70].
Various investigators have also reported a failure to thrive in a few pediatric patients [21,36].
3.5. Immunological Characterization (Table 5)
As previously mentioned, several authors have reported additional immunological abnormalities in patients initially identified with SIgMD.
IgG subclass deficiency has been reported in a subset of SIgMD patients with a rate between 25% [19] and 42% [71], particularly IgG4 subclass deficiency [72,73].
T cell number and function are normal in most SIgMD patients [34,35,37,74]. However, alterations in subsets of patients have been reported [36]. In their study, Lucuab-Fergurur et al. reported that 10% of their adult subjects had reduced CD3+ T cells, 30% had reduced CD4+ T helper cells, and 2% had reduced CD8+ T cells [68].
B cells are normal in most SIgMD patients. However, low B cells have been reported in a few patients with SIgMD [43,68,74]. Non-switched memory B cell values were significantly lower in a subgroup of SIgMD patients than in healthy controls [75,76]. Mensen et al. found reduced class-switched memory B cells, but naïve B cells were normal in their cohort [77].
The lymphocytic proliferation response to mitogens and antigens is maintained in most patients with SIgMD [78]. Raziuddin et al. reported CD4+ T cell deficiency and defective interleukin 2 receptor expression and production by the patient’s peripheral blood lymphocytes in response to mitogenic stimulation. Thus, impaired T cell function may be responsible for IgM-deficient antibody production [79]. Yamasaki et al. previously described a reduction in the proliferative response of patient B cells to Staphylococcus aureus Cowan strain I (SAC). Considering in vitro immunoglobulin production, IgM production could be normal [74] or reduced. At the same time, IgG and IgA have been reported to be normal (using a T cell-independent culture system) [35], as reported by Karsh et al. [32]. Considering the response to polysaccharides and proteic antigens, Lucuab et al. reported an unprotected/impaired anti-Streptococcus pneumoniae antibody in 47% of patients and an inadequate response to tetanus toxoid in up to 12% of patients in their cohort [36,68].
3.6. Therapeutic Interventions (Table 6)
No conclusive data on the correct therapeutic management of SIgMD are available. Although, for most SIgMD patients, Ig replacement therapy (IgGRT) is not required [71], it may be recommended for patients with significantly associated antibody deficiency or recurrent or severe infections [5,80]. It is also interesting to observe that Goldstein et al. reported that IgGRT in patients with SIgMID and bronchiectasis might lead to a reduced risk of pulmonary infection with consequently better infection control and a preventive role in further progressive bronchiectasis [81]. Notably, it is conceivable that IgM-enriched immunoglobulin replacement therapy with biologically active IgM can potentially prevent bacterial respiratory infections. However, further studies must be conducted to determine the potential role of IgM-enriched IgRT [82]. Prophylactic antibiotics and the prompt treatment of febrile illness are crucial. Adult patients receive more frequent antibiotics and/or IgGRT [20,68] compared to the pediatric population [21]. Vaccines, including pneumococcal and meningococcal vaccines, should be given as scheduled for healthy populations. However, antibody response may be decreased, and conjugate vaccines may require repeated doses to provide protection (if any). However, considering the immunological abnormalities associated with SIgMD, the impairment of T cell function should be excluded before the administration of attenuated vaccines.
3.7. Prognosis
Although, in some infants, SIgMD may be transient [43], insufficient evidence is available on the prognosis of this condition. In particular, no conclusive data have been collected regarding the course of the disease and the possible evolution of SIgMD to common variable immunodeficiency (CVID) [83].
4. Discussion
SIgMD has been recently included in the IEI classification by the IUIS Expert Committee [1,2,3]. However, as recently reported by the US National Institutes of Health (https://rarediseases.info.nih.gov/diseases/12547/selective-igm-deficiency (accessed on 29 May 2023)), our understanding of this condition requires improvement.
The definition of SIgMD is still controversial. Although specific ESID criteria are available, some authors propose that SIgMD should be defined without the exclusion of the IgG subclass deficiency, alterations in T cell subset numbers and functions, and impaired response to the vaccine in order to better understand the different clinical and immunological phenotypes that lie behind the diagnostic term “IgM deficiency.” Indeed, as demonstrated by Janssen et al., only a limited number of SIgMD-diagnosed patients reported in the literature adhere to the ESID criteria [7]. From a clinical perspective, according to the Jeffrey Modell Foundation’s “Four stages of testing” [84], patients usually undergo quantitative IgG-, IgM-, and IgA-level dosage as the first step in the immunological workup. From the available evidence, it is clear that patients exhibiting absent/reduced serum IgM levels after this first stage of testing should undergo a complete immunological workup to identify other possibly associated immunological abnormalities.
According to our literature review, the main clinical manifestations are represented by infections, especially respiratory infections, followed by allergic manifestations [85,86] and autoimmune diseases [20,21,68,87]. Neoplastic diseases have been described with a non-conclusive association with SIgMD.
Considering the probably pathogenetic defects associated with SIgMD reported in the literature, some of them are of particular interest, namely BTK gene mutation [29,30], BLNK gene mutation [30], BAFFR deficiency due to TNFRSF13C gene mutation [31], and Igμ deficiency [33]. Some of these are also causes of agammaglobulinemia. In this category of IEI, we have the BTK deficiency X-linked agammaglobulinemia (XLA), the autosomal recessive μ heavy chain deficiency, and BLNK deficiency [2]. These conditions are characterized by severe bacterial infections, a severe reduction in all serum immunoglobulin isotypes, and profoundly decreased or absent B cells (CD19+ less than 2%) [88]. These features are partially shared with SIgMD, reinforcing the etiopathogenetic hypothesis that it could be determined by a B-cell intrinsic defect. BAFF receptor deficiency due to TNFRSF13C gene mutation is one the IEI showing a CVID phenotype (defined as an age-specific reduction in the serum concentrations of IgG in combination with low levels of IgA and/or IgM and a poor or absent response to immunizations and/or absent isohemagglutinins and/or low switched B cells (<70 percent of age-related normal value) and the absence of profound T cell immunodeficiency and absence of any other defined immunodeficient state [89,90]) characterized by low IgG and IgM serum levels with a variable clinical expression [3]. This consideration strengthens the possible evolution of SIgMD to CVID [83] and the need for a close follow-up for all patients with SIgMD.
Regarding the immunological characterization, according to ESID criteria [4] and Janssen et al.’s definition [7,46], “truly” selective SIgMD has been defined as repeatedly decreased serum IgM levels, normal levels of serum IgG, IgA, IgG subclasses, and normal vaccination responses and the exclusion of T cell defects through the absence of clinical signs suggesting a T cell defect. However, as previously mentioned, several additional immunological abnormalities have been reported in the literature. These findings strengthen the above recommendation for a complete immunological workup in patients with absent/reduced serum IgM levels.
No conclusive data regarding the course of the disease, its potential therapeutic management, and the possible evolution of SIgMD to CVID are available. The most important forms of therapeutic intervention are represented by measures to prevent infections. Both inactivated vaccines (e.g., vaccines against Tetanus, Diphtheria, Pertussis), and live attenuated vaccines (e.g., vaccines against measles, rubeola, mumps, chickenpox, and Rotavirus) are safe in minor antibody deficiencies; thus, they are also safe in SIgMD. On the other hand, in antibody deficiency disorder, the response to active immunization can be highly variable in patients with antibody defects, from cases with an adequate response, as in healthy subjects, to cases in which it is reduced or even absent [91], as reported above. As reported by Ko et al., it is plausible that it could depend on B-memory cell values for polysaccharide antigens in CVID [92]. Because there are several reports of patients with SIgMD with associated T cell defects before administering live attenuated vaccines, it is recommended to check the lymphocyte subsets to inoculate these vaccines safely. In particular, the following immunological parameters are requested: CD4+ T cells ≥500 cells/μL, CD8+ T cells ≥200 cells/μL, and normal T cell response to mitogen (The Center for Disease Control and Prevention recommends even higher CD4+ levels in children below the 6 years of age, namely, CD4+ ≥1000 cells/μL for those between 1 and 6 years of age and ≥1500 cells/μL for those under one year of age)[93].
An accurate extended follow-up (clinical and immunological) in Immunology Clinics, even in asymptomatic patients, is crucial in order to evaluate the evolution of SIgMD in children and adults (persistent or transitory) and identify the possible progression to CVID or other well-defined immunodeficiencies, with a specific focus on patients who showed immune abnormalities in addition to low IgM [20,94].
5. Conclusions
Our overall understanding of SIgMD is still extremely limited, especially with respect to the pediatric population. This review evaluated the evidence regarding SIgMD to clarify the condition’s etiopathogenetic and pathophysiologic mechanisms, its main clinical manifestations and immunological features, and its potential treatment options in order to define present and future needs. Further studies, including prospective longitudinal studies with regular immunological evaluation and, if possible, genetic testing, are required to understand the disease trajectories and increase our understanding of the molecular mechanisms underlying SIgMD to facilitate a better clinical, immunological, and prognostic characterization of the condition and therefore develop tailored therapeutic management strategies.
References
- Picard, C.; Bobby Gaspar, H.; Al-Herz, W.; Bousfiha, A.; Casanova, J.-L.; Chatila, T.; Crow, Y.J.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J. Clin. Immunol. 2018, 38, 96–128. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1473–1507. [Google Scholar] [CrossRef] [PubMed]
- Bousfiha, A.; Moundir, A.; Tangye, S.G.; Picard, C.; Jeddane, L.; Al-Herz, W.; Rundles, C.C.; Franco, J.L.; Holland, S.M.; Klein, C.; et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J. Clin. Immunol. 2022, 42, 1508–1520. [Google Scholar] [CrossRef] [PubMed]
- ESID—European Society for Immunodeficiencies. Available online: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria (accessed on 25 March 2023).
- Gupta, S.; Gupta, A. Selective IgM Deficiency—An Underestimated Primary Immunodeficiency. Front. Immunol. 2017, 8, 1056. [Google Scholar] [CrossRef]
- Lozano, R.; Marin, R.; Santacruz, M.-J.; Pascual, A. Selective Immunoglobulin M Deficiency Among Clozapine-Treated Patients: A Nested Case-Control Study. Prim. Care Companion CNS Disord. 2015, 17. [Google Scholar] [CrossRef]
- Janssen, L.M.A.; Macken, T.; Creemers, M.C.W.; Pruijt, J.F.M.; Eijk, J.J.J.; de Vries, E. Truly selective primary IgM deficiency is probably very rare. Clin. Exp. Immunol. 2018, 191, 203–211. [Google Scholar] [CrossRef]
- Arani, M.H.; Razavizadeh, M.; ArefNezhad, R.; Motedayyen, H. Selective immunoglobulin M deficiency in a patient with celiac disease and recurrent pneumonia. Clin. Case Rep. 2021, 9, 158–163. [Google Scholar] [CrossRef]
- Gupta, S.; Gupta, A. Defining Primary Selective IgM Deficiency. J. Clin. Immunol. 2019, 39, 350–352. [Google Scholar] [CrossRef]
- Hobbs, J.R.; Milner, R.D.; Watt, P.J. Gamma-M deficiency predisposing to meningococcal septicaemia. Br. Med. J. 1967, 4, 583–586. [Google Scholar] [CrossRef]
- Thong, Y.H.; Maxwell, G.M. Primary selective deficiency of immunoglobulin M. Aust. N. Z. J. Med. 1978, 8, 436–438. [Google Scholar] [CrossRef]
- Kutukculer, N.; Gulez, N. The outcome of patients with unclassified hypogammaglobulinemia in early childhood. Pediatr. Allergy Immunol. 2009, 20, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Núñez, R.M. Primary immunodeficiency in Colombian children. Allergol. Immunopathol. 1988, 16, 273–275. [Google Scholar]
- Datta, U.; Kaur, K.J.; Kumar, L.; Walia, B.N.; Mehta, S.; Mehta, S.K.; Sehgal, S. Immunoglobulin deficiency. Indian Pediatr. 1993, 30, 461–467. [Google Scholar] [PubMed]
- Ni, J.; Zhang, J.; Chen, Q.; Chen, Y.; Liu, J. The epidemiology and clinical features of selective immunoglobulin M deficiency: A single-center study in China. J. Clin. Lab. Anal. 2020, 34, e23289. [Google Scholar] [CrossRef] [PubMed]
- Louis, A.G.; Gupta, S. Primary Selective IgM Deficiency: An Ignored Immunodeficiency. Clinic Rev. Allergy Immunol. 2014, 46, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.T.; Nordby, G.L. Human serum immunoglobulin concentrations: Prevalence of immunoglobulin deficiencies. J. Allergy Clin. Immunol. 1975, 55, 35–48. [Google Scholar] [CrossRef]
- Entezari, N.; Adab, Z.; Zeydi, M.; Saghafi, S.; Jamali, M.; Kardar, G.A.; Pourpak, Z. The prevalence of Selective Immunoglobulin M Deficiency (SIgMD) in Iranian volunteer blood donors. Hum. Immunol. 2016, 77, 7–11. [Google Scholar] [CrossRef]
- Goldstein, M.F.; Goldstein, A.L.; Dunsky, E.H.; Dvorin, D.J.; Belecanech, G.A.; Shamir, K. Selective IgM immunodeficiency: Retrospective analysis of 36 adult patients with review of the literature. Ann. Allergy Asthma Immunol. 2006, 97, 717–730. [Google Scholar] [CrossRef]
- Caka, C.; Cimen, O.; Kahyaoğlu, P.; Tezcan, İ.; Cagdas, D. Selective IgM deficiency: Follow-up and outcome. Pediatr. Allergy Immunol. 2021, 32, 1327–1334. [Google Scholar] [CrossRef]
- Goldstein, M.F.; Goldstein, A.L.; Dunsky, E.H.; Dvorin, D.J.; Belecanech, G.A.; Shamir, K. Pediatric Selective IgM Immunodeficiency. J. Immunol. Res. 2008, 2008, e624850. [Google Scholar] [CrossRef]
- Janssen, L.M.A.; Reijnen, I.C.G.M.; Milito, C.; Edgar, D.; Chapel, H.; de Vries, E. Protocol for the unclassified primary antibody deficiency (unPAD) study: Characterization and classification of patients using the ESID online Registry. PLoS ONE 2022, 17, e0266083. [Google Scholar] [CrossRef]
- Celmeli, F.; Turkkahraman, D.; Cetin, Z.; Mihci, E.; Yegin, O. Selective IgM deficiency in a boy with ring chromosome 18. J. Investig. Allergol. Clin. Immunol. 2014, 24, 442–444. [Google Scholar] [PubMed]
- Seidel, M.G.; Duerr, C.; Woutsas, S.; Schwerin-Nagel, A.; Sadeghi, K.; Neesen, J.; Uhrig, S.; Santos-Valente, E.; Pickl, W.F.; Schwinger, W.; et al. A novel immunodeficiency syndrome associated with partial trisomy 19p13. J. Med. Genet. 2014, 51, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Inoue, C.N.; Tanaka, Y.; Tabata, N. Acne conglobata in a long-term survivor with trisomy 13, accompanied by selective IgM deficiency. Am. J. Med. Genet. Part A 2017, 173, 1903–1906. [Google Scholar] [CrossRef] [PubMed]
- Al-Herz, W.; McGeady, S.J.; Gripp, K.W. 22q11.2 deletion syndrome and selective igm deficiency: An association of a common chromosomal abnormality with a rare immunodeficiency. Am. J. Med. Genet. Part A 2004, 127A, 99–100. [Google Scholar] [CrossRef]
- Kung, S.-J.; Gripp, K.W.; Stephan, M.J.; Fairchok, M.P.; McGeady, S.J. Selective IgM deficiency and 22q11.2 deletion syndrome. Ann. Allergy Asthma Immunol. 2007, 99, 87–92. [Google Scholar] [CrossRef]
- Ozawa, T.; Kondo, N.; Motoyoshi, F.; Kasahara, K.; Orii, T. Dna Mutation Induced in the Sequence Upstream of the Secreted Myu C-Terminal Coding Sequence by Ultraviolet Irradiation in the Cell Line of Bloom’s Syndrome. Int. J. Immunogenet. 1995, 22, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.-M.; Chang, J.-M.; Wang, I.-F.; Chang, W.-C.; Hwang, D.-Y.; Chen, H.-C. Atypical X-linked agammaglobulinaemia caused by a novel BTK mutation in a selective immunoglobulin M deficiency patient. BMC Pediatr. 2013, 13, 150. [Google Scholar] [CrossRef]
- Geier, C.B.; Sauerwein, K.M.T.; Leiss-Piller, A.; Zmek, I.; Fischer, M.B.; Eibl, M.M.; Wolf, H.M. Hypomorphic Mutations in the BCR Signalosome Lead to Selective Immunoglobulin M Deficiency and Impaired B-cell Homeostasis. Front. Immunol. 2018, 9, 2984. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02984 (accessed on 30 May 2023). [CrossRef]
- Smulski, C.R.; Eibel, H. BAFF and BAFF-Receptor in B Cell Selection and Survival. Front. Immunol. 2018, 9, 2285. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2018.02285 (accessed on 20 May 2023). [CrossRef]
- Karsh, J.; Watts, C.S.; Osterland, C.K. Selective immunoglobulin M deficiency in an adult: Assessment of immunoglobulin production by peripheral blood lymphocytes in vitro. Clin. Immunol. Immunopathol. 1982, 25, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Ozawa, T.; Kato, Y.; Motoyoshi, F.; Kasahara, K.; Kameyama, T.; Orii, T. Reduced secreted μ mRNA synthesis in selective IgM deficiency of Bloom’s syndrome. Clin. Exp. Immunol. 1992, 88, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Inaba, M.; Kuribayashi, K.; Masuda, T.; Kanoh, T.; Uchino, H. Selective IgM deficiency in adults: Phenotypically and functionally altered profiles of peripheral blood lymphocytes. Clin. Exp. Immunol. 1987, 68, 630–637. [Google Scholar] [PubMed]
- Yamasaki, T. Selective IgM Deficiency: Functional Assessment of Peripheral Blood Lymphocytes in vitro. Intern. Med. 1992, 31, 866–870. [Google Scholar] [CrossRef]
- De la Concha, E.G.; Garcia-Rodriguez, M.C.; Zabay, J.M.; Laso, M.T.; Alonso, F.; Bootello, A.; Fontan, G. Functional assessment of T and B lymphocytes in patients with selective IgM deficiency. Clin. Exp. Immunol. 1982, 49, 670–676. [Google Scholar]
- Louis, A.G.; Agrawal, S.; Gupta, S. Analysis of subsets of B cells, Breg, CD4Treg and CD8Treg cells in adult patients with primary selective IgM deficiency. Am. J. Clin. Exp. Immunol. 2016, 5, 21–32. [Google Scholar]
- Kasahara, T.D.M.; Bento, C.A.D.M.; Gupta, S. Phenotypic analysis of T follicular helper and T follicular regulatory cells in primary selective IgM deficiency. Hum. Immunol. 2020, 81, 625–633. [Google Scholar] [CrossRef]
- Inoue, T.; Okumura, Y.; Shirahama, M.; Ishibashi, H.; Kashiwagi, S.; Okubo, H. Selective partial IgM deficiency: Functional assessment of T and B lymphocytesin vitro. J. Clin. Immunol. 1986, 6, 130–135. [Google Scholar] [CrossRef]
- Matsushita, S.; Inoue, T.; Okubo, H. A Case of Selective IgM Deficiency: Isotype-specific Suppressor T Lymphocytes. Jpn. J. Med. 1984, 23, 149–151. [Google Scholar] [CrossRef]
- Gupta, S.; Agrawal, S.; Gollapudi, S.; Kubagawa, H. FcμR in human B cell subsets in primary selective IgM deficiency, and regulation of FcμR and production of natural IgM antibodies by IGIV. Hum. Immunol. 2016, 77, 1194–1201. [Google Scholar] [CrossRef]
- Fallon, K.E. Inability to train, recurrent infection, and selective IgM deficiency. Clin. J. Sport. Med. 2004, 14, 357–359. [Google Scholar] [CrossRef]
- Ozen, A.; Baris, S.; Karakoc-Aydiner, E.; Ozdemir, C.; Bahceciler, N.N.; Barlan, I.B. Outcome of hypogammaglobulinemia in children: Immunoglobulin levels as predictors. Clin. Immunol. 2010, 137, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Barker, A.F.; Craig, S.; Bardana, E.J. Humoral immunity in bronchiectasis. Ann. Allergy 1987, 59, 179–182. [Google Scholar] [PubMed]
- Dhir, V.; Sagar, V.; Aggarwal, A.; Rawat, A.; Singhal, M. An unusual cause of recurrent pneumonia in adults. Lung Indian 2014, 31, 296–298. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.M.A.; van Hout, R.W.N.M.; de Vries, E.; Pignata, C.; Cirillo, E.; Arkwright, P.D.; Lougaris, V.; Buckland, M.; Garcia-Prat, M.; D’Hebron, V.; et al. Challenges in investigating patients with isolated decreased serum IgM: The SIMcal study. Scand. J. Immunol. 2019, 89, e12763. [Google Scholar] [CrossRef]
- Phuphuakrat, A.; Ngamjanyaporn, P.; Nantiruj, K.; Luangwedchakarn, V.; Malathum, K. Selective IgM deficiency in an adult presenting with Streptococcus pneumoniae septic arthritis. J. Microbiol. Immunol. Infect. 2016, 49, 150–153. [Google Scholar] [CrossRef]
- Indian Pediatrics-Editorial. Available online: https://www.indianpediatrics.net/sep2005/sep-961-962.htm (accessed on 22 February 2023).
- Ross, I.N.; Thompson, R.A. Severe selective IgM deficiency. J. Clin. Pathol. 1976, 29, 773–777. [Google Scholar] [CrossRef]
- Hong, R.; Gupta, S. Selective immunoglobulin M deficiency in an adult with Streptococcus pneumoniae sepsis and invasive aspergillosis. J. Investig. Allergol. Clin. Immunol. 2008, 18, 214–218. [Google Scholar]
- Kampitak, T. Selective immunoglobulin M deficiency in a patient with refractory giardiasis. J. Investig. Allergol. Clin. Immunol. 2010, 20, 358–360. [Google Scholar]
- Moise, A.; Nedelcu, F.; Toader, M.; Sora, S.; Tica, A.; Ferastraoaru, D.; Constantinescu, I. Primary immunodeficiencies of the B Lymphocyte. J. Med. Life 2010, 3, 60–63. [Google Scholar]
- Hassanein, H.A.; Elbadry, M.I. Selective immunoglobulin M deficiency in an adult with miliary tuberculosis: A clinically interesting coexistence. A case report and review of the literature. Int. J. Mycobacteriol. 2016, 5, 106–110. [Google Scholar] [CrossRef]
- Consonni, F.; Chiti, N.; Ricci, S.; Venturini, E.; Canessa, C.; Bianchi, L.; Lippi, F.; Montagnani, C.; Giovannini, M.; Chiappini, E.; et al. Unbalanced serum immunoglobulins in clinical subtypes of pediatric tuberculosis disease. Front. Pediatr. 2022, 10, 908963. [Google Scholar] [CrossRef]
- Mayumi, M.; Yamaoka, K.; Tsutsui, T.; Mizue, H.; Doi, A.; Matsuyama, M.; Ito, S.; Shinomiya, K.; Mikawa, H. Selective immunoglobulin M deficiency associated with disseminated molluscum contagiosum. Eur. J. Pediatr. 1986, 145, 99–103. [Google Scholar] [CrossRef]
- Yocum, M.W.; Strong, D.M.; Chusid, M.J.; Lakin, J.D. Selective immunoglobulin M (IgM) deficiency in two immunodeficient adults with recurrent staphylococcal pyoderma. Am. J. Med. 1976, 60, 486–494. [Google Scholar] [CrossRef]
- Belgemen, T.; Suskan, E.; Dogu, F.; Ikinciogullari, A. Selective Immunoglobulin M Deficiency Presenting with Recurrent Impetigo: A Case Report and Review of the Literature. Int. Arch. Allergy Immunol. 2009, 149, 283–288. [Google Scholar] [CrossRef]
- Kıratlı, H.K.; Akar, Y. Multiple recurrent hordeola associated with selective IgM deficiency. J. Am. Assoc. Pediatr. Ophthalmol. Strabismus 2001, 5, 60–61. [Google Scholar] [CrossRef]
- Campochiaro, C.; Atay, S.; Clark, K.E.N.; Ong, V.; Denton, C.P. Autoimmunity and immunodeficiency at the crossroad: Autoimmune disorders as the presenting feature of selective IgM deficiency. BMJ Case Rep. CP 2019, 12, e223180. [Google Scholar] [CrossRef]
- Takeuchi, T.; Nakagawa, T.; Maeda, Y.; Hirano, S.; Sasaki-Hayashi, M.; Makino, S.; Shimizu, A. Functional Defect of B Lymphocytes in a Patient with Selective IgM Deficiency Associated with Systemic Lupus Erythematosus. Autoimmunity 2001, 34, 115–122. [Google Scholar] [CrossRef]
- Kimura, S.; Tanigawa, M.; Nakahashi, Y.; Inoue, M.; Yamamura, Y.; Kato, H.; Sugino, S.; Kondo, M. Selective IgM Deficiency in a Patient with Hashimoto’s Disease. Intern. Med. 1993, 32, 302–307. [Google Scholar] [CrossRef]
- Antar, M.; Lamarche, J.; Peguero, A.; Reiss, A.; Cole, S. A case of selective immunoglobulin M deficiency and autoimmune glomerulonephritis. Clin. Exp. Nephrol. 2008, 12, 300–304. [Google Scholar] [CrossRef]
- Sano, A.; Inoue, J.; Kakazu, E.; Ninomiya, M.; Iwata, T.; Morosawa, T.; Takai, S.; Nakamura, T.; Masamune, A. Acute-onset Autoimmune Hepatitis in a Patient with Selective Immunoglobulin M Deficiency. Intern. Med. 2019, 58, 2185–2190. [Google Scholar] [CrossRef]
- Oh, J.; McGarry, D.; Peppers, B.; Hostoffer, R. Selective IgM deficiency associated with adult-onset Still disease. Ann. Allergy Asthma Immunol. 2018, 120, 444–446. [Google Scholar] [CrossRef]
- Makay, B.; Ünsal, E.; Anal, Ö.; Güneş, D.; Men, S.; Çakmakçı, H.; Özer, E. Chronic recurrent multifocal osteomyelitis in a patient with selective immunoglobulin M deficiency. Rheumatol. Int. 2009, 29, 811–815. [Google Scholar] [CrossRef]
- Agarwal, A.; Sharma, S.; Airun, M. Symptomatic Primary Selective IgM Immunodeficiency—B Lymphoid Cell Defect in Adult Man with Secondary HLH Syndrome. J. Assoc. Physicians India 2016, 64, 91–93. [Google Scholar]
- Saini, S.; Dettore, A.J.; Bhambhani, K.J.; Buck, S.; Poulik, J.; Savasan, S. Selective IgM Deficiency in CD30+ Cutaneous Lymphoproliferative Disorder. J. Pediatr. Hematol./Oncol. 2011, 33, e156. [Google Scholar] [CrossRef]
- Lucuab-Fegurgur, D.L.; Gupta, S. Comprehensive clinical and immunological features of 62 adult patients with selective primary IgM deficiency. Am. J. Clin. Exp. Immunol. 2019, 8, 55–67. [Google Scholar] [PubMed]
- Narsai, T.; Su, H.; Braxton, D.; Gupta, S. Collagenous Gastritis in Primary Selective IgM Deficiency: Transition to EBV+ Gastric Adenocarcinoma. Case Rep. Immunol. 2021, 2021, 5574944. [Google Scholar] [CrossRef]
- Gupta, S.; Agrawal, S. IgAλ monoclonal gammopathy of undetermined significance (MGUS) associated with primary selective IgM deficiency. Am. J. Clin. Exp. Immunol. 2019, 8, 37–46. [Google Scholar]
- Chovancova, Z.; Kralickova, P.; Pejchalova, A.; Bloomfield, M.; Nechvatalova, J.; Vlkova, M.; Litzman, J. Selective IgM Deficiency: Clinical and Laboratory Features of 17 Patients and a Review of the Literature. J. Clin. Immunol. 2017, 37, 559–574. [Google Scholar] [CrossRef]
- Bolia, R.; Misra, D.P.; Aggarwal, A.; Srivastava, A. Paediatric selective IgM deficiency and IgG4 deficiency: An extremely unusual association. Case Rep. 2014, 2014, bcr2014204769. [Google Scholar] [CrossRef]
- Ideura, G.; Agematsu, K.; Komatsu, Y.; Hatayama, O.; Yasuo, M.; Tsushima, K.; Hanaoka, M.; Koizumi, T.; Fujimoto, K.; Kubo, K. Selective IgM deficiency accompanied with IgG4 deficiency, dermal complications and a bronchial polyp. Allergol. Int. 2008, 57, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Endoh, M.; Kaneshige, H.; Tomino, Y.; Nomoto, Y.; Sakai, H.; Arimori, S. Selective IgM deficiency: A case study. Tokai J. Exp. Clin. Med. 1981, 6, 327–331. [Google Scholar] [PubMed]
- Celiksoy, M.H.; Yildiran, A. A comparison of B cell subsets in primary immune deficiencies that progress with antibody deficiency and age-matched healthy children. Allergol. Immunopathol. 2016, 44, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Cipe, F.E.; Doğu, F.; Güloğlu, D.; Aytekin, C.; Polat, M.; Biyikli, Z.; Ikincioğullari, A. B-cell subsets in patients with transient hypogammaglobulinemia of infancy, partial IgA deficiency, and selective IgM deficiency. J. Investig. Allergol. Clin. Immunol. 2013, 23, 94–100. [Google Scholar]
- Mensen, A.; Krause, T.; Hanitsch, L.G.; Meisel, C.; Kleint, M.E.; Volk, H.-D.; Na, I.-K.; Scheibenbogen, C. Altered B-cell subsets and functional B-cell defects in selective IgM deficiency. Clin. Immunol. 2015, 161, 96–102. [Google Scholar] [CrossRef]
- Moffitt, J.E.; Guill, M.F.; Wray, B.B.; Brown, D.A.; Peacocke, N.W.; Ades, E.W. Effect of interleukin-2 and mitogen on in vitro immunoglobulin production by peripheral blood lymphocytes from patients with selective IgM deficiency. Ann. Allergy 1988, 61, 424–427. [Google Scholar]
- Raziuddin, S.; Bilal, N.; Benjamin, B. Transient T-cell abnormality in a selective IgM-immunodeficient patient with Brucella infection. Clin. Immunol. Immunopathol. 1988, 46, 360–367. [Google Scholar] [CrossRef]
- Patel, S.S.; Fergeson, J.E.; Glaum, M.C.; Lockey, R.F. Symptomatic Primary Selective Immunoglobulin M Deficiency with Nonprotective Pneumococcal Titers Responsive to Subcutaneous Immunoglobulin Treatment. Int. Arch. Allergy Immunol. 2016, 170, 138–140. [Google Scholar] [CrossRef]
- Goldstein, M.F.; Hilditch, G.J.; Dvorin, D.J.; Belecanech, G.A. Immunoglobulin replacement for selective IgM immunodeficiency, bronchiectasis, and asthma. Ann. Allergy Asthma Immunol. 2016, 116, 172–173. [Google Scholar] [CrossRef]
- Langereis, J.D.; van der Flier, M.; de Jonge, M.I. Limited Innovations After More Than 65 Years of Immunoglobulin Replacement Therapy: Potential of IgA- and IgM-Enriched Formulations to Prevent Bacterial Respiratory Tract Infections. Front. Immunol. 2018, 9, 1925. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2018.01925 (accessed on 9 May 2023). [CrossRef]
- Narsai, T.; Gupta, S. Progression of primary selective immunoglobulin M deficiency to common variable immunodeficiency. Ann. Allergy Asthma Immunol. 2021, 126, 723–724. [Google Scholar] [CrossRef] [PubMed]
- Educational Materials/Library/JMF. Available online: https://info4pi.org/library/educational-materials/ (accessed on 22 June 2023).
- Guill, M.F.; Brown, D.A.; Ochs, H.D.; Pyun, K.H.; Moffitt, J.E. IgM deficiency: Clinical spectrum and immunologic assessment. Ann. Allergy 1989, 62, 547–552. [Google Scholar] [PubMed]
- Kaufman, H.; Hobbs, J. Immunoglobulin deficiencies in an atopic population. Lancet 1970, 296, 1061–1063. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Gupta, A. Chapter 6—Autoimmunity and autoimmune diseases in primary selective IgM deficiency. In Translational Autoimmunity; Rezaei, N., Ed.; Translational Immunology; Academic Press: Cambridge, MA, USA, 2022; Volume 3, pp. 129–139. ISBN 978-0-323-85415-3. [Google Scholar]
- Gaspar, H.B.; Conley, M.E. Early B cell defects. Clin. Exp. Immunol. 2000, 119, 383–389. [Google Scholar] [CrossRef]
- Bonilla, F.A.; Barlan, I.; Chapel, H.; Costa-Carvalho, B.T.; Cunningham-Rundles, C.; de la Morena, M.T.; Espinosa-Rosales, F.J.; Hammarström, L.; Nonoyama, S.; Quinti, I.; et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J. Allergy Clin. Immunol. Pract. 2016, 4, 38–59. [Google Scholar] [CrossRef]
- Seidel, M.G.; Kindle, G.; Gathmann, B.; Quinti, I.; Buckland, M.; van Montfrans, J.; Scheible, R.; Rusch, S.; Gasteiger, L.M.; Grimbacher, B.; et al. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J. Allergy Clin. Immunol. Pract. 2019, 7, 1763–1770. [Google Scholar] [CrossRef]
- Martire, B.; Azzari, C.; Badolato, R.; Canessa, C.; Cirillo, E.; Gallo, V.; Graziani, S.; Lorenzini, T.; Milito, C.; Panza, R.; et al. Vaccination in immunocompromised host: Recommendations of Italian Primary Immunodeficiency Network Centers (IPINET). Vaccine 2018, 36, 3541–3554. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.; Radigan, L.; Cunningham-Rundles, C. Immune competence and switched memory B cells in common variable immunodeficiency. Clin. Immunol. 2005, 116, 37–41. [Google Scholar] [CrossRef]
- American Academy of Pediatrics; Committee on Infectious Diseases. Red Book: 2015 Report of the Committee on Infectious Diseases; American Academy of Pediatrics: Itasca, IL, USA, 2015. [Google Scholar]
- Cunningham-Rundles, C. Common variable immune deficiency: Case studies. Hematology 2019, 2019, 449–456. [Google Scholar] [CrossRef]